Laboratorio di Modeling Molecolare e Drug Design (MMDDLab)
(stanze Q165_107A, Q165_102)
Responsabile Prof. Roberta Galeazzi
Attività di ricerca 
Il Laboratorio di Modellistica Molecolare e Drug Design (MMDDLab) è un laboratorio di ricerca specializzato in tecniche di chimica computazionale, modellistica molecolare applicate nella ricerca in ambito della chimica organica, bioorganica biomolecolare e chimico-biologica. L’attività del MMDDLab comprende principalmente: la progettazione molecolare assistita al computer (in silico) con particolare interesse verso le strategie razionali “ligand-based drug design” e “structure-based drug design” (CADD, Computer Aided Drug Design), lo studio delle interazioni ligando-recettore, substrato-proteina, studio computazionale di meccanismi di reazione enzimatici, studi relativi a struttura e folding di peptidi e proteine, modelling di proteine per omologia, relazioni struttura-attività, progettazione di materiali nanostrutturati, studio dinamico strutturale di bilayer lipidici a composizione mista applicati al Gene e Drug Delivery, simulazioni di dinamica molecolare nel bilayer lipidico di sistemi proteici quali recettori di membrana e proteine sensore. Altre competenze del laboratorio suddetto: Caratterizzazione chimico-fisica degli aggregati supramolecolari lipidici e correlazione delle loro caratteristiche strutturali con la capacità di complessare farmaci o acidi nucleici da veicolare all' interno delle cellule.
Progetti e tematiche di ricerca
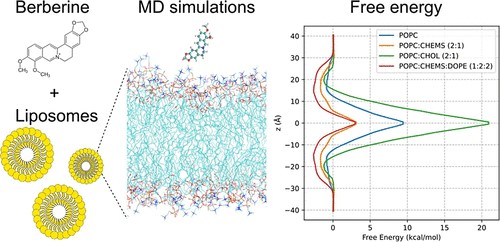
- Progettazione e Studio Strutturale/Dinamico di Bilayers a composizione mista costituenti liposomi come Nanovettori per il Gene e Drug Delivery
-
Progettazione, sintesi e Studio Strutturale/Dinamico di Bilayers a composizione mista costituenti liposomi Nanovettori per il Gene e Drug Delivery (collaborazione con Laboratorio di Bionanotecnologie)
• Liposomi funzionalizzati coordinanti cationi e DNA
• Liposomi sensibili al pH
• Liposomi coniugati con molecole antiossidanti in grado di svolgere un effetto protettivo in differenti condizioni di stress ossidativo.

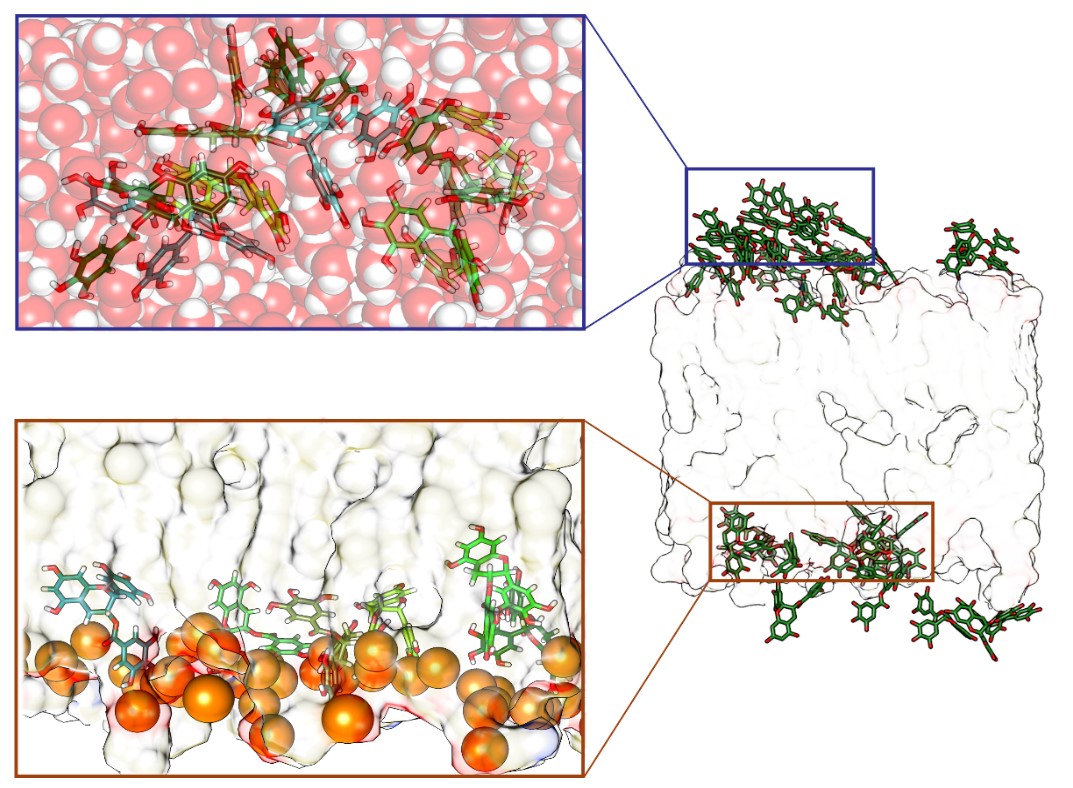
- Predizione della permeazione di membrane cellulari di molecole bioattive mediante metodiche computazionali avanzate
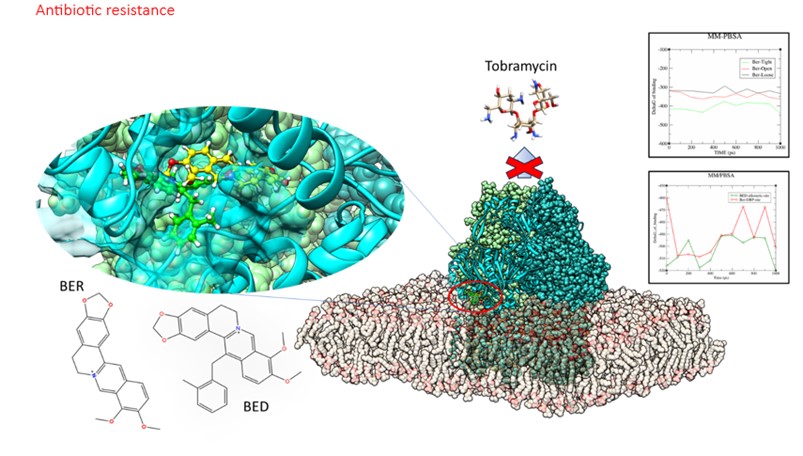
- Sviluppo e identificazione di inibitori di pompe di efflusso batteriche (EPIs) per il superamento delle resistenze in Pseudomonas Aeruginosa con target MexY e MexB delle pompe di efflusso MexXY-OprM e MexAB-OprM
- Meccanismi molecolari del Quorum sensing alla base della formazione del biofilm; identificazione di inibitori selettivi per LasR, RhIR in P. aeruginosa.
-
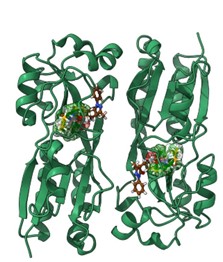
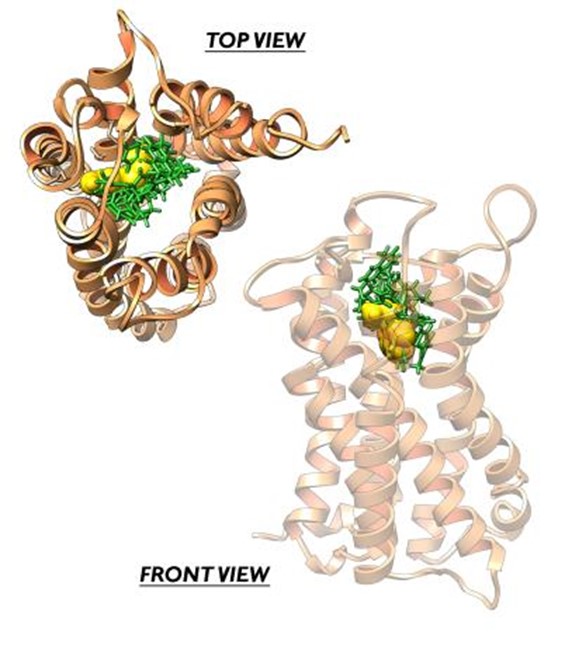
- Dinamica del Meccanismo di attivazione/inibizione dei recettori serotoninergici GPCRs (i.e 5-HT2C, 5-HT1A). Affinità di binding e polimorfismi genetici. Meccanismo di traslazione e trasduzione del segnale biologico e dimerizzazione (Coarse Grained/full atom MD); influenza degli ioni nel pathway di attivazione; design di agonisti ad attività selettiva.
-
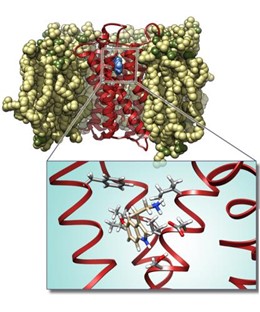
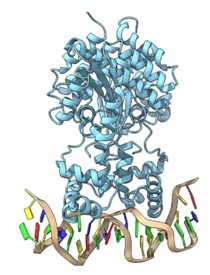
- Meccanismi molecolari dell’attivazione e design di agonisti per il recettore nucleare orfano Nurr1 per il trattamento del morbo di Parkinson
-
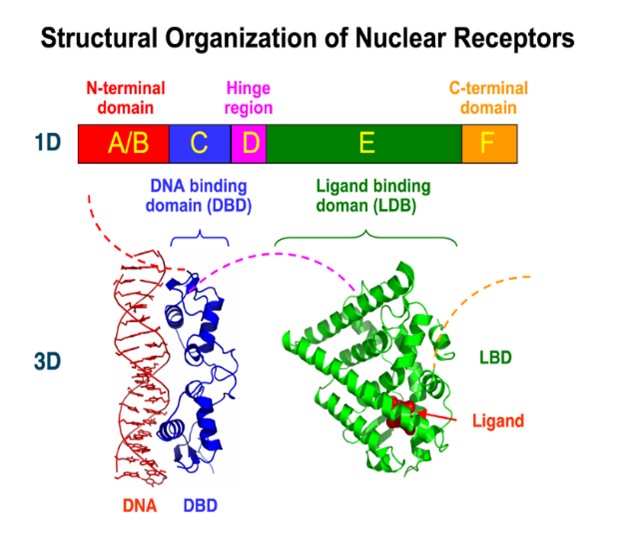
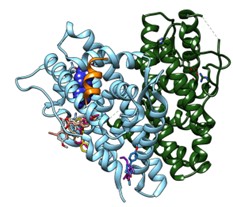
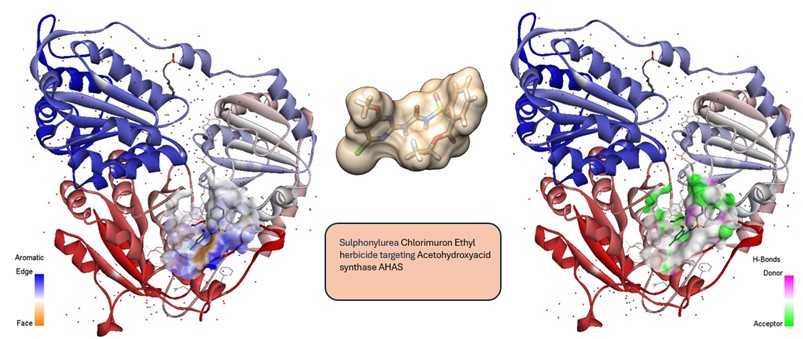
- Design razionale e sviluppo di pesticidi naturali per una agricoltura biosostenibile
- Design di Molecular Imprinted Polymers (MIPs) per lo sviluppo di anticorpi sintetici selettivi per target macromolecolari specifici (Her2, Spike protein)
-
- Campi di Ricerca
-
• Progettazione di nuove molecole attive a target enzimatico o recettoriale come potenziali agenti farmacologici mediante l’uso di tecniche di modellistica molecolare e il “de novo” drug design. Sulla base dell’identificazione, infatti, delle interazioni molecolari responsabili delle associazioni substrato-target recettoriale è possibile modificare la struttura “ad hoc” al fine di massimizzare tali interazioni responsabili della affinità e/o crearne delle altre aggiuntive. Tale procedura può portare all’identificazione di nuovi composti lead, la cui attività può essere aumentata mediante cicli ripetuti di structure-based optimization utilizzando metodi avanzati di docking molecolare e virtual screening finalizzati a prevedere l’affinità delle nuove molecole. Nei casi in cui la struttura 3D del target recettoriale non sia nota è possibile ricorrere alle tecniche del comparative molecular docking combinato a simulazioni di dinamica molecolare per costruire il modello strutturale del target macromolecolare.
• Studio meccanicistico dei meccanismi molecolari alla base di importanti processi biologici. Particolare attenzione è rivolta alle proteine di membrana, soprattutto GPCRs e ai recettori nucleari. Le metodiche computazionali e di simulazione molecolare permettono di comprendere tali meccanismi, la cui conoscenza è fondamentale nel design di nuove molecole bioattive.
• Simulazione e design del bilayer lipidico in dinamica molecolare costituisce una nuova frontiera nell’ambito della modellistica molecolare sia per la complessità del sistema sia per l’importanza scientifica; difatti, una corretta rappresentazione a livello molecolare di tali sistemi offre la possibilità di interpretare e progettare molti dati sperimentali, come proprietà di materiali nano strutturati, fino ad ora mai caratterizzati e permette lo studio della permeabilità di membrane biologiche.
La Dinamica Molecolare sia unbiased che enanched sampling è sicuramente la tecnica piu’ utilizzata nel LMMDLab, oltre al consensus docking e Hight Throuput virtual Screening, in quanto grazie alla sua versatilità può essere impiegata per la risoluzione di problematiche di vario tipo, che vanno dalla semplice analisi conformazionale e previsione di strutture secondarie (per esempio in peptidomimetici , foldameri) alla simulazione di processi di associazione ligando-proteina, di folding/unfolding proteico, di ricostruzione del bilayer lipidico e dell’organizzazione dei sistemi proteina-bilayer. Pertanto tale metodica risulta particolarmente utile nel valutare la stabilità dei complessi di associazione substrato-recettore e nello studio dei recettori di membrana nel loro ambiente nativo (bilayer lipidico).

Le attività di ricerca del laboratorio MMDD sono integrate e aperte a collaborazioni con altri gruppi di ricerca a livello nazionale ed internazionale che permettono una validazione sperimentale dei modelli previsionali messi a punto (come sintesi, attività biologica e altre proprietà)
Nell’ambito delle competenze di ricerca del gruppo, si inserisce la collaborazione scientifica costante con la start up innovativa in ambito nutraceutico Biosolving s.r.l (www.biosolving.com) (Dott. Luca Massaccesi)

Strumentazione
- Strumentazione scientifica
-
Laboratory facilities: N.1 Worktations GPU based with NVIDIA GEFORCE RTX 3070 (winblu i7 Expert Z490 10TH) and N.1 RTX 4070 SUPER (AMD RYZEN 9 7900 (3.7 GHz) - SSD nVme 1TBRAM - DDR5 64GB - Raffreddamento a liquido - GEFORCE RTX 4070 SUPER 12GB; three ubuntu worstations NVIDIA GPU based i5/i7 intel. Software Licenses for docking and Dynamics Packages (Autodock, GROMACS, AMBER, Autodock Vina GPU, VMD/NAMD, G16).
HPC access to HPC (High Performance Computing) server at DISVA
HPC access to HPC cineca ISCRA project class C and B - Strumentazione didattica
-
Il laboratorio MMDDLab è dotato di n.4 postazioni dotate di Workstation/Linux based per lo svolgimento delle attività di tesi sperimentali di laurea magistrale e attività di tirocinio.
- Software in dotazione presso il laboratorio
-
Il laboratorio dispone di un'ampia varietà di software di modellazione molecolare. Di seguito è riportato un elenco di alcuni programmi utilizzato con una breve descrizione. Per informazioni dettagliate, visitare il link di ogni singolo programma.
AMBER - AMBER, by David Case at The Scripps Research Institute and collaborators, is the collective name for a suite of programs that allow users to carry out molecular dynamics simulations, particularly on biomolecules. http://ambermd.org/
Gaussian 09 (G09) - A suite of programs to perform semi-empirical and ab initio molecular orbital calculations on Linux/UNIX based machines (CINECA-ISCRA). http://www.gaussian.com/
UCSF Chimera is a highly extensible program for interactive visualization and analysis of molecular structures and related data, including density maps, supramolecular assemblies, sequence alignments, docking results, trajectories, and conformational ensembles. http://plato.cgl.ucsf.edu/chimera/
Autodock- MGLTools; AutodockVina-GPU: suite of automated docking tools. It is designed to predict how small molecules, such as substrates or drug candidates, bind to a receptor of known 3D structure. http://autodock.scripps.edu/
GROMACS- versatile package to perform molecular dynamics, i.e. simulate the Newtonian equations of motion for systems with hundreds to millions of particles. It is primarily designed for biochemical molecules like proteins and lipids that have a lot of complicated bonded interactions, but since GROMACS is extremely fast at calculating the nonbonded interactions (that usually dominate simulations) many groups are also using it for research on non-biological systems, e.g. polymers. http://www.gromacs.org/
VMD/NAMD: NAMD, is a parallel molecular dynamics code designed for high-performance simulation of large biomolecular systems. Based on Charm++ parallel objects, NAMD scales to hundreds of processors on high-end parallel platforms and tens of processors on commodity clusters using gigabit ethernet. NAMD uses the popular molecular graphics program VMD for simulation setup and trajectory analysis, but is also file-compatible with AMBER, CHARMM, and X-PLOR. NAMD is distributed free of charge with source code. http://www.ks.uiuc.edu/Research/namd/
MODELLER is used for homology or comparative modeling of protein three-dimensional structures. http://salilab.org/modeller/
 |
|
 |
 |
Staff
|
tel. 071 2204724
|
 |
|
Dott. Luca Massacesi
collaboratore Biosolving s.r.l.
|
 |
|
||
|
Collaborazione continua con Laboratorio di Bionanotecnologie del SIMAU: Dr. Michela Pisani (SIMAU), responsabile Unità di caratterizzazione biofisica e chimico-fisica
|
|||||||