Molecular Modeling and Drug Design (MMDDLab) Lab
(room Q165_107A, Q165_102)
Responsible Prof. Roberta Galeazzi
Research Activities
The Molecular Modeling and Drug Design Laboratory (MMDDLab) is a research laboratory specialized in computational chemistry techniques and molecular modeling, applied to research in the fields of organic, bioorganic, biomolecular, and chemical biology. MMDDLab's activities primarily include: computer-aided molecular design, with a particular focus on rational strategies such as ligand-based drug design and structure-based drug design (CADD, Computer-Aided Drug Design); the study of ligand-receptor and substrate-protein interactions; computational investigation of enzymatic reaction mechanisms; studies on the structure and folding of peptides and proteins; homology modeling of proteins; structure-activity relationships; design of nanostructured materials; structural and dynamic studies of mixed-composition lipid bilayers applied to gene and drug delivery; and molecular dynamics simulations of protein systems, such as membrane receptors and sensor proteins, within lipid bilayers.
Additional expertise of the laboratory includes the physicochemical characterization of lipid polymolecular aggregates and the correlation between their structural features and their ability to complex drugs or nucleic acids for intracellular delivery.
Research Projects and Topics
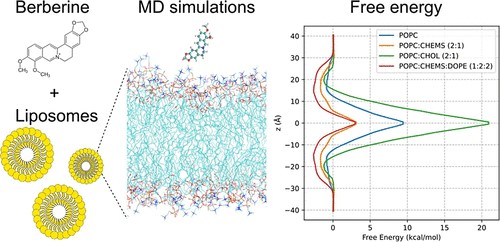
- Design and Structural/Dynamics of Mixed-Composition Bilayers Forming Liposomes as Nanocarriers for Gene and Drug Delivery
-
Progettazione, sintesi e Studio Strutturale/Dinamico di Bilayers a composizione mista costituenti liposomi Nanovettori per il Gene e Drug Delivery (collaborazione con Laboratorio di Bionanotecnologie)
• Liposomi funzionalizzati coordinanti cationi e DNA
• Liposomi sensibili al pH
• Liposomi coniugati con molecole antiossidanti in grado di svolgere un effetto protettivo in differenti condizioni di stress ossidativo.

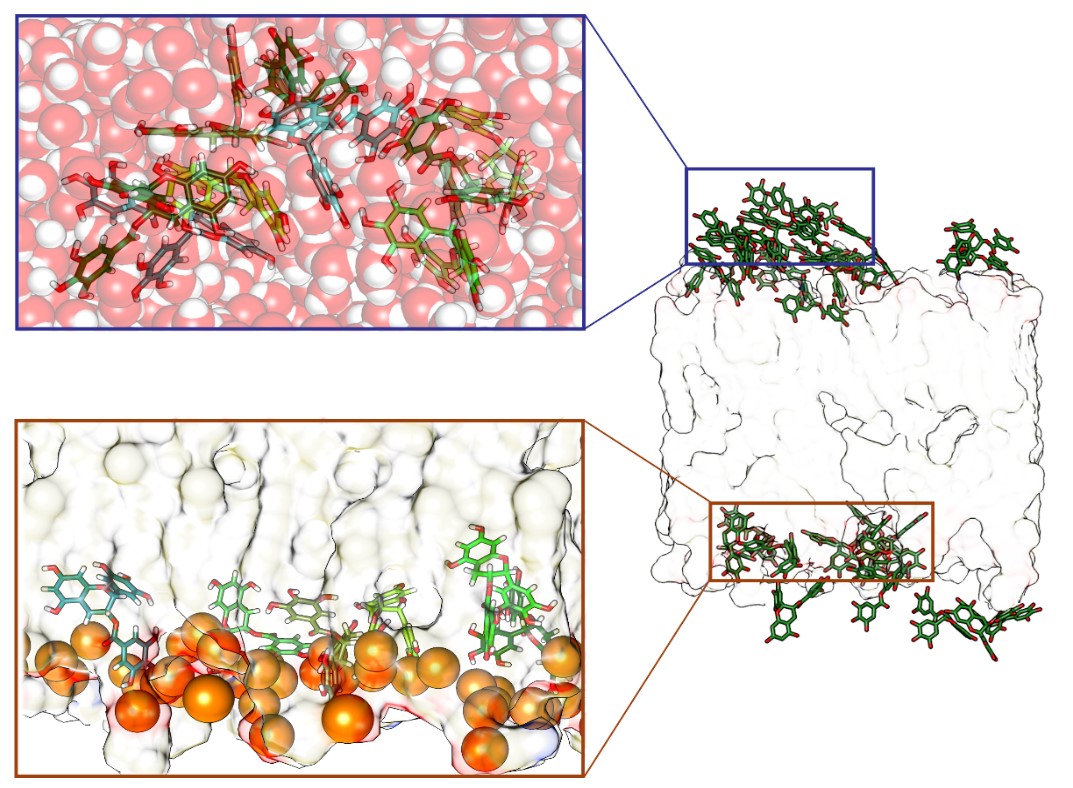
- Prediction of the membrane permeability of bioactive molecules using advanced computational methods and molecular dynamics simualations
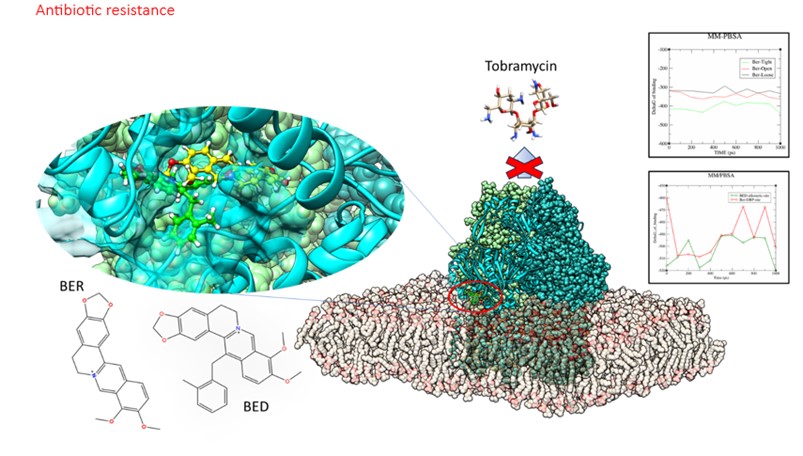
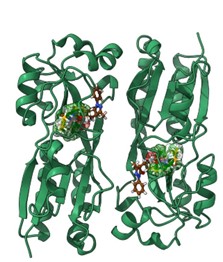
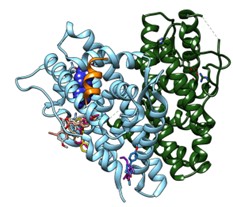
- Development and identification of bacterial efflux pump inhibitors (EPIs) to overcome resistance in Pseudomonas aeruginosa, targeting MexY and MexB, inner macromlecular component of the efflux pumps MexXY-OprM and MexAB-OprM
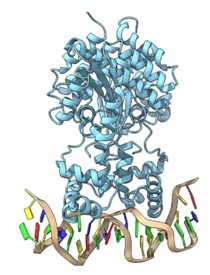
- Molecular mechanisms of quorum sensing underlying biofilm formation; identification of selective inhibitors for LasR and RhIR in P. aeruginosa
-
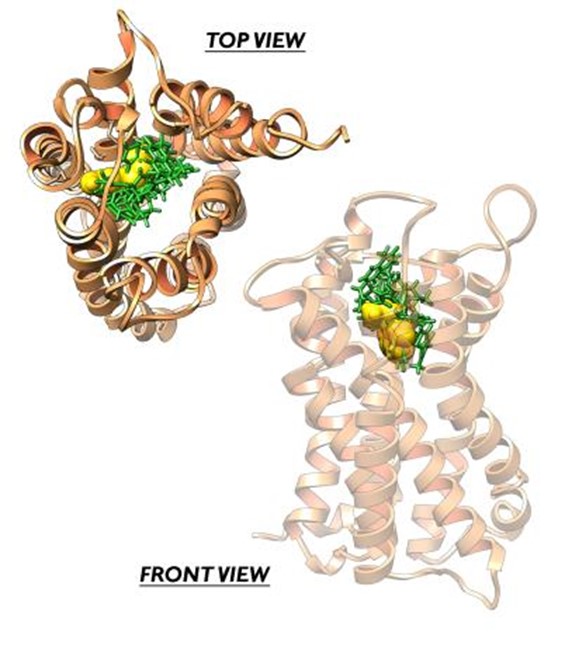
- Dynamics of activation/inhibition mechanisms of serotonergic GPCRs (e.g., 5-HT2C, 5-HT1A); binding affinity and genetic polymorphisms. Mechanism of biological signal translation and transduction, and receptor dimerization (Coarse-Grained and full-atom molecular dynamics simulations).Ions influence in the activation pathway of this class of receptors; design of selective agonists with anxiolytic activity
-
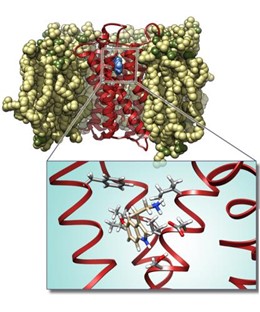
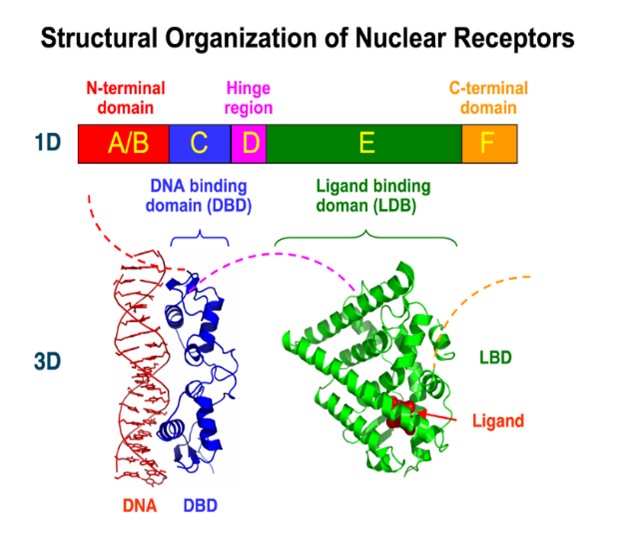
- Molecular mechanisms of activation and agonist design for the Nurr1 receptor for the treatment of Parkinson’s disease
-
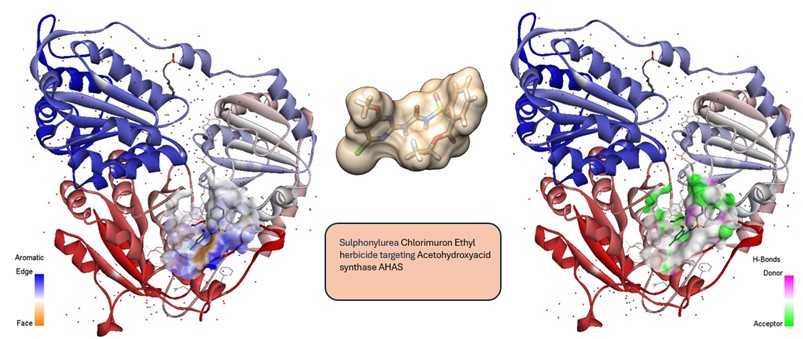
- Rational design and development of natural pesticides for sustainable agriculture
- Design of Molecularly Imprinted Polymers (MIPs) for the development of synthetic antibodies selectively targeting specific macromolecular targets (Her2, Spike protein)
-
- Research Areas
-
The research activity of the Molecular Modeling and Drug Design Laboratory (MMDDLab) focuses on:
• The design of new molecules targeting enzymes or receptors as potential pharmacological agents, using molecular modeling techniques and de novo drug design approaches. Based on the identification of molecular interactions responsible for substrate–receptor target binding, molecular structures can be modified ad hoc to maximize these interactions, thus enhancing affinity and/or to introduce new favorable interactions. This strategy can lead to the identification of new lead compounds whose activity can be optimized through iterative cycles of structure-based optimization, using advanced molecular docking methods and virtual screening to predict binding affinity of the designed molecules. When the 3D structure of the receptor target is unknown, comparative molecular docking techniques combined with molecular dynamics simulations can be employed to build the structural model of the macromolecular target.
• Mechanistic study of molecular processes underlying important biological phenomena. Special attention is given to membrane proteins, particularly GPCRs and nuclear receptors. Computational modeling and molecular simulation techniques allow for a deep understanding of these mechanisms, which are essential for the rational design of new bioactive molecules.
• The simulation of lipid bilayers using molecular dynamics represents a new frontier in molecular modeling, due to the complexity and scientific significance of these systems. Accurate molecular-level representation of such systems enables interpretation and design of experimental data, including properties of nanostructured materials, which until now have remained uncharacterized.Molecular dynamics simulations (both unbiased and enhanced sampling) are among the most widely used techniques at MMDDLab, alongside consensus docking and high-throughput virtual screening. Thanks to its versatility, molecular dynamics is employed to address various challenges, from conformational analysis and secondary structure prediction (e.g., in peptidomimetics or foldamers), to the simulation of ligand–protein association processes, protein folding/unfolding, and the reconstruction and organization of lipid bilayers and protein–bilayer systems. This technique is especially useful for evaluating the stability of substrate–receptor complexes and for studying membrane receptors in their native lipid bilayer environment.

MMDDLab’s research activities are integrated and open to collaborations with other research groups at both national and international levels, allowing for experimental validation of the predictive models developed (e.g., synthesis, biological activity, and other properties).
Within the group’s research expertise, there is also a long-standing collaboration with the innovative nutraceutical start-up Biosolving s.r.l. (www.biosolving.com) (Dott. Luca Massaccesi).

Equipment
- Scientific Equipment
-
Laboratory facilities include:
• 1 GPU-based workstation with NVIDIA GEFORCE RTX 3070 (Winblu i7 Expert Z490 10th Ge
• 1 GPU based workstation with RTX 4070 SUPER (AMD RYZEN 9 7900 @ 3.7 GHz, 1TB NVMe SSD, 64GB DDR5 RAM, liquid cooling, GEFORCE RTX 4070 SUPER 12GB
• 3 Ubuntu-based workstations with NVIDIA GPU, Intel i5/i7 processors.
• Software licenses for docking and molecular dynamics packages: Autodock, GROMACS, AMBER, Autodock Vina GPU, VMD/NAMD, Gaussian 16 (G16).
• Access to High Performance Computing (HPC) servers at DISVA.
• Access to CINECA HPC through ISCRA Class C and B projects. - Educational Equipment
-
The MMDDLab is equipped with 4 workstation/Linux setups dedicated to experimental Master’s thesis work and internship activities
- Software Available in the Laboratory
-
The laboratory has a wide range of molecular modeling software. Below is a list of some of the programs used, with a brief description. For more detailed information, please refer to the respective software’s official websites
AMBER - AMBER, by David Case at The Scripps Research Institute and collaborators, is the collective name for a suite of programs that allow users to carry out molecular dynamics simulations, particularly on biomolecules. http://ambermd.org/
Gaussian 09 (G09) - A suite of programs to perform semi-empirical and ab initio molecular orbital calculations on Linux/UNIX based machines (CINECA-ISCRA). http://www.gaussian.com/
UCSF Chimera is a highly extensible program for interactive visualization and analysis of molecular structures and related data, including density maps, supramolecular assemblies, sequence alignments, docking results, trajectories, and conformational ensembles. http://plato.cgl.ucsf.edu/chimera/
Autodock- MGLTools; AutodockVina-GPU: suite of automated docking tools. It is designed to predict how small molecules, such as substrates or drug candidates, bind to a receptor of known 3D structure. http://autodock.scripps.edu/
GROMACS- versatile package to perform molecular dynamics, i.e. simulate the Newtonian equations of motion for systems with hundreds to millions of particles. It is primarily designed for biochemical molecules like proteins and lipids that have a lot of complicated bonded interactions, but since GROMACS is extremely fast at calculating the nonbonded interactions (that usually dominate simulations) many groups are also using it for research on non-biological systems, e.g. polymers. http://www.gromacs.org/
VMD/NAMD: NAMD, is a parallel molecular dynamics code designed for high-performance simulation of large biomolecular systems. Based on Charm++ parallel objects, NAMD scales to hundreds of processors on high-end parallel platforms and tens of processors on commodity clusters using gigabit ethernet. NAMD uses the popular molecular graphics program VMD for simulation setup and trajectory analysis, but is also file-compatible with AMBER, CHARMM, and X-PLOR. NAMD is distributed free of charge with source code. http://www.ks.uiuc.edu/Research/namd/
MODELLER is used for homology or comparative modeling of protein three-dimensional structures. http://salilab.org/modeller/
 |
|
 |
 |
Staff
|
tel. 071 2204724
|
 |
|
Dott. Luca Massacesi
collaboratore Biosolving s.r.l.
|
 |
|||
|
Collaborazione continua con Laboratorio di Bionanotecnologie del SIMAU: Dr. Michela Pisani (SIMAU), responsabile Unità di caratterizzazione biofisica e chimico-fisica
|
|||||||